The Larimore Lab
The Role of Endosomal Trafficking in Neuronal Development
The Larimore lab has focused on endosomal vesicle trafficking disruptions in neurodevelopment disorders from 2012 to present. Our research has utilized several mouse models of autism, Rett syndrome, and schizophrenia to study development. Over the next five to ten years, we plan to continue our work on exploring the role of endosomal trafficking in development - exploring cellular phenotypes, receptor trafficking, and ultimately behavior at several key developmental time points using the SHANK3B +/1 mouse model of autism. A grant has been submitted to help support this work and is currently under review.
Vesicles that traffic through the endosomal pathway are derived from two distinct donor membranes – the Golgi complex or the plasma membrane. Those vesicles originating from the Golgi complex are trafficked directly to the plasma membrane or to an early endosome for further sorting, unless they are mis-folded, in which case they traffic from the Golgi directly to the late endosome. There are specific ADP-ribosylation factors (Arfs), ArfGTPase associated proteins (GAPs), coat proteins, and associated coat proteins that are specific for this anterograde trafficking (Bonifacino and Glick, 2004). The Arf GAPs AGAP1, AGAP2, and AGAP5 have been implicated in several neurodevelopmental diseases (including Autism Spectrum Disorder, schizophrenia, and Trisomy 23), but their exact role in typical neurodevelopment remains unknown. AGAP1, specifically interacts with two other vesicle complexes that are also implicated in several neurodevelopmental disorders: the coat complex AP-3 and the Biogenesis of Lysosome Related Organelles -1 (BLOC1) complex to form vesicles on endosomes.
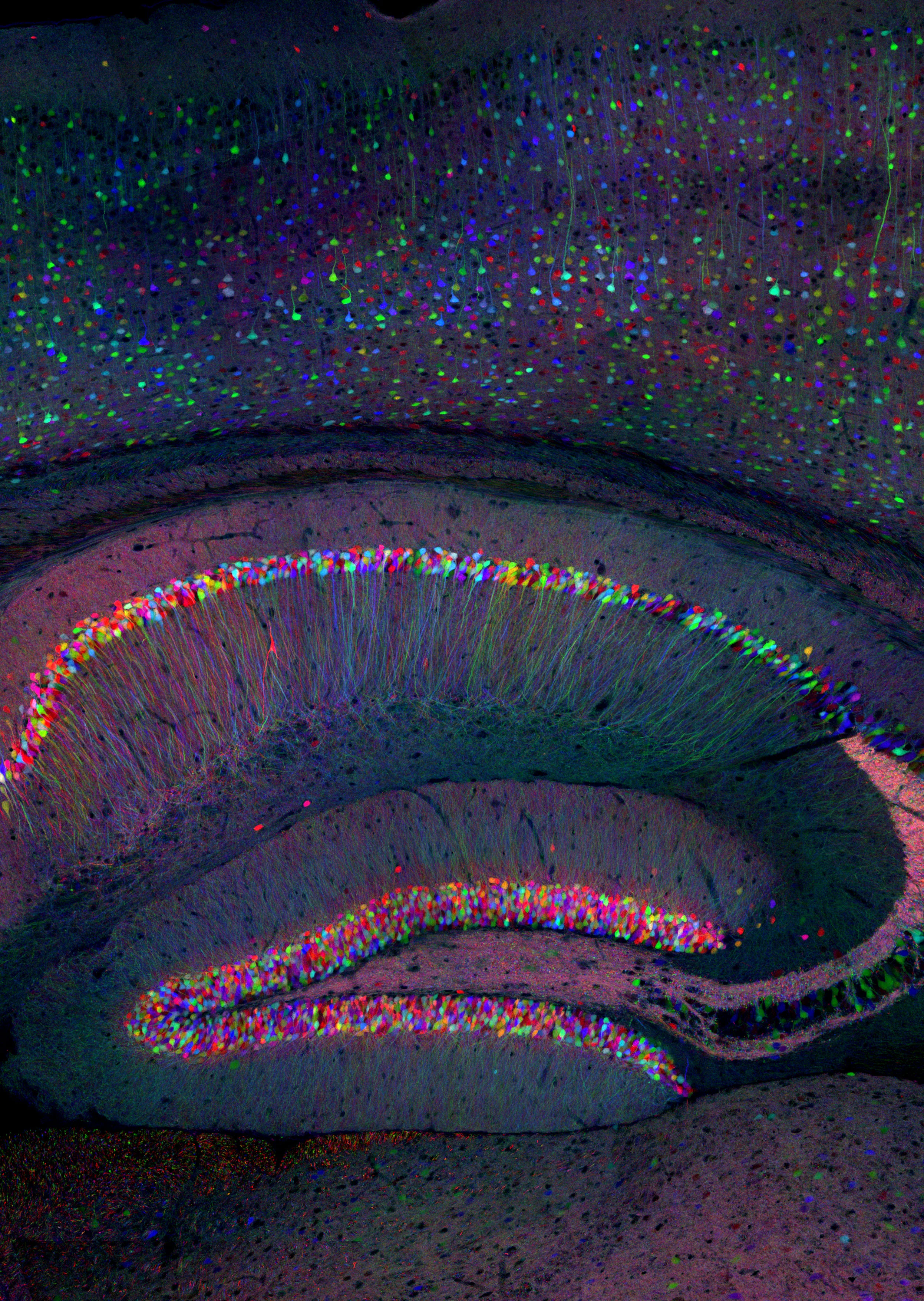
Of the neurodevelopmental disorders that Arf GAPs have been genetically implicated in, one common phenotype is altered abilities in learning and memory. In order for dendritic spines to maintain proper morphology (stubby Type 1 or mushroom Type II) required for synaptic plasticity, recycling endosomes must be fully functional within the dendrite. When endosomes are blocked/unable to traffic, spines shifted from a stable phenotype to an unstable phenotype (thin/filamentous. Type III) (Park et al., 2006a). Spine density has been reported in the SHANK3B -/- mouse, but not in a sex dependent manner and not in the heterozygote. Further, endosomal recycling of receptors has been demonstrated necessary to maintain a mobile pool of AMPA receptors that are necessary for LTP (Park et al., 2004), the cellular correlation for learning and memory. Additionally, endosomal trafficking is also necessary for the proper trafficking of key receptors to the spines in dendrites in the hippocampus. AMPA receptors require endosomal trafficking in order to maintain the proper number of receptors inserted in the postsynaptic density (PSD).
We are starting in fall 2024 by examining the protein levels and localization of excitatory/inhibitory markers in the hippocampus of the SHANK3B +/1 mouse model as well as key trafficking markers. We are starting our studies at a 12 week time point to pair the data with observed behavioral changes. After we complete the studies at 12 weeks, we will explore younger times in development.
Below is one of our abstracts submitted and accepted for presentation at the Society for Neuroscience:
Dendritic morphology and dendritic spine receptor insertion is necessary for typical neuronal development and synaptic formation. Previous studies have proposed that aberrant connectivity among neurons underlies autism phenotypes, and that altered connectivity is a result, in part, of altered dendritic spine volume and density of patients with autism. Postsynaptic densities (PSD) composition requires proper endosomal trafficking at the level of the recycling endosome as well as other scaffolding proteins to be present. The SHANK family of proteins are responsible for synapse formation and synaptic plasticity at glutamatergic synapses. SHANK3B codes for key PSD proteins that are part of the glutamate receptor protein complex that physically links ionotropic NMDA receptors to metabotropic mGlu5 receptors, a linkage necessary for induction of plasticity. Using immunoblotting, immunohistochemistry, and qt-PCR we are exploring how AGAP1-dependent endosomal trafficking kinetics and endosomal protein levels are altered in SHANK3B +/- mice. We are also exploring how those alterations regulate receptor trafficking, receptor localization in the PSD, and spine morphology.\
Research Publications since earning tenure in Spring 2018.
Susan Cordero Romero, Ruvimbo Dzvurumi, Alexia Crockett, Alexandra Lombardo, Jhodi Webster, Samantha Hatcher, Alix Wagner, Diana Ghebrezadik, Asiya Abawari, Camryn Smith, Lauren Neal, Yommi Tadesse, M. Beauchamp, Stacey B.B. Dutton and Jennifer L. Larimore. Endosomal trafficking is disrupted in Neurodevelopmental Disorders. J Brain Research. 2021 January.
Frank Y Lee, Jennifer Larimore, Victor Faundez, Esteban C Dell'Angelica, Cristina A Ghiani. Sex-dimorphic Effects of Biogenesis of Lysosome-Related Organelles complex-1 Deficiency on Mouse Perinatal Brain Development. J Neuroscience Research. 2020 May 20. doi: 10.1002/jnr.24620.